Protein Misfolding and Aggregation
Proteins are the working molecules of the cell. For them to function properly, they must be made with the right sequence, folded into the right shape, transported to the right place, and eventually broken down when no longer needed. Protein aggregation is the process by which proteins lose their proper structure and begin to stick together, forming abnormal clumps inside or outside of cells.
While small amounts of misfolded protein are normal and constantly handled by the cell, problems arise when this system becomes overwhelmed. Misfolded proteins can accumulate and form aggregates that interfere with normal cellular function. In many neurodegenerative diseases, these aggregates are a defining feature and are often used to classify the disease itself. Beyond just accumulating, these misfolded proteins can also induce other proteins to misfold which can aggravate or spread the disease.
Protein aggregation is not just a byproduct of disease. In many cases, it plays an active role in driving cellular dysfunction. Across conditions such as Alzheimer’s disease, Parkinson’s disease, ALS/FTD, and Huntington’s disease, aggregation represents a breakdown of the cell’s ability to maintain protein homeostasis, often called proteostasis.
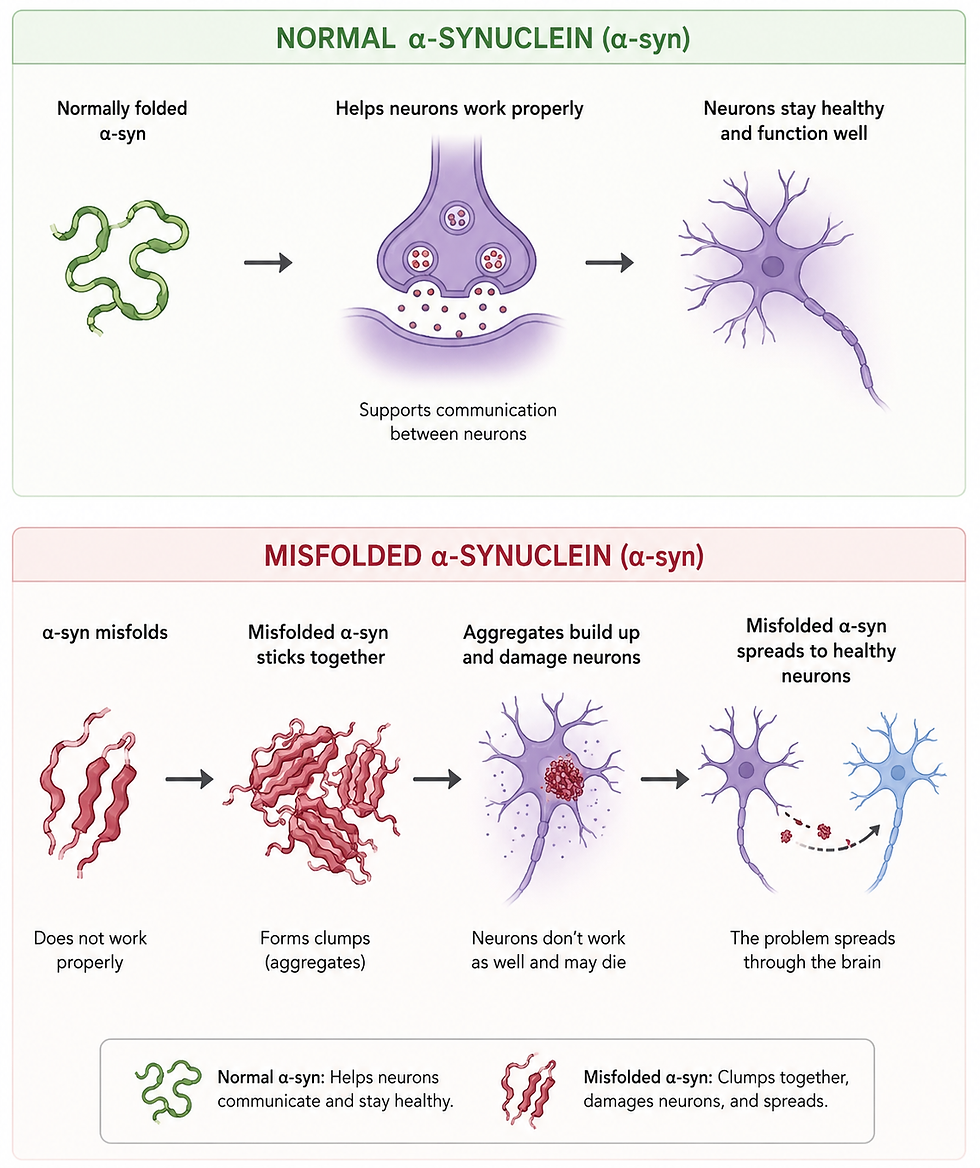
Normal Biology
In healthy cells, protein quality is tightly controlled through a system known as the proteostasis network. This includes molecular chaperones that help proteins fold correctly, as well as degradation systems that remove damaged or unnecessary proteins. Proteins are synthesized in the ribosome and must fold into specific three-dimensional shapes to function properly. Molecular chaperones assist in this process, preventing misfolding and helping refold damaged proteins when possible.
If proteins cannot be properly folded, they are targeted for degradation. There are two major systems responsible for handling this: the ubiquitin-proteasome system which marks short-live or damaged proteins with a molecule called ubiquitin that marks them for degradation and the autophagy-lysosome system which handles protein aggregates and damaged organelles. Together, these systems maintain a balance between protein production, folding, and clearance. This balance is especially important in neurons, which must maintain protein integrity over long periods without dividing.
Dysfunction
Protein aggregation occurs when the proteostasis network is overwhelmed or impaired. This can happen due to mutations in specific proteins (chaperones and those involved in degradation), increased production of aggregation-prone proteins, or stressors such as oxidative damage.
Misfolded proteins can expose normally hidden regions that make them sticky, leading them to bind to each other and form oligomers and larger aggregates. These aggregates can accumulate in different parts of the cell like the cytoplasm, nucleus, or extracellular space.
Interestingly, the most toxic species are often not the large visible aggregates, but smaller intermediate forms, called oligomers. These can interact with cellular components, disrupt membranes, and interfere with signaling.
As aggregation progresses, it can impair the very systems responsible for clearing misfolded proteins. The proteasome and autophagy pathways become less effective, leading to further accumulation. This creates a continuous cycle of proteostasis failure.
Disease Connections
Protein aggregation is a defining feature of many neurodegenerative diseases, though the specific proteins involved differ.
In Alzheimer’s disease, amyloid-beta forms extracellular plaques, while tau forms intracellular tangles. These aggregates are closely associated with synaptic dysfunction and neuronal loss. However, emerging research calls into questions whether it is the aggregates themselves or an overactive immune response to them that is to blame.
In Parkinson’s disease, the protein alpha-synuclein aggregates into structures known as Lewy bodies. These aggregates are found in vulnerable dopaminergic neuronal populations and are thought to disrupt cellular function.
In ALS/FTD, proteins such as TDP-43 and FUS mislocalize and form aggregates, often accompanied by loss of their normal function. This dual effect is particularly important in these diseases.
In Huntington’s disease, the mutant huntingtin protein forms intracellular aggregates that cause with neuronal dysfunction, particularly in the striatum.
In TBI/CTE, repetitive injury is associated with accumulation of abnormal tau, linking protein aggregation to trauma-related neurodegeneration.
Across these diseases, protein aggregation provides a unifying framework: different proteins, but similar underlying failure of proteostasis.
Molecular Consequences
At the molecular level, protein aggregation disrupts multiple cellular systems.
One key effect is loss of normal protein function. When proteins misfold and aggregate, they are no longer able to perform their intended roles. This is particularly important for proteins like TDP-43, which regulate RNA processing.
Another effect can be toxic gain of function. Aggregated proteins can interact abnormally with membranes, organelles, and other proteins, disrupting cellular processes. For example, oligomeric species can form pores in membranes or interfere with synaptic function.
Protein aggregation also places stress on the endoplasmic reticulum (which is where proteins are made and folded) and activates the unfolded protein response (UPR). While initially protective, chronic activation of these pathways can contribute to cell death.
There are also strong links between protein aggregation and mitochondrial dysfunction, oxidative stress, and impaired autophagy. Aggregates can physically or functionally interfere with these systems, creating a network of interconnected stress responses.
Therapeutic Targeting
Targeting protein aggregation has been a major focus of drug development, particularly in diseases like Alzheimer’s and Parkinson’s.
One strategy is to reduce the production of aggregation-prone proteins. For example, therapies that lower amyloid-beta or mutant huntingtin levels aim to reduce the burden of toxic proteins.
Another approach is to enhance protein clearance, either by boosting proteasome activity or stimulating autophagy. This aims to restore the cell’s ability to remove aggregates.
There is also interest in stabilizing protein folding using small molecules or chaperone-based therapies, as well as preventing aggregation by interfering with protein-protein interactions with a class of molecules called foldamers.
However, clinical success has been mixed. One challenge is that aggregation often begins years before symptoms appear, meaning interventions may need to occur earlier. Another is that simply removing aggregates does not always reverse existing damage.
Research Direction
Current research is focused on better understanding the dynamics of protein aggregation and identifying the most toxic species.
One major question is the role of oligomers versus large aggregates. Increasing evidence suggests that smaller aggregates may be more harmful than the larger deposits seen on pathology.
Another area of interest is the concept of prion-like spread, where misfolded proteins can propagate their abnormal structure to other proteins and spread through neural networks. This may help explain how disease progresses through the brain, and especially how Parkinson’s might be influenced by the gut.
Researchers are also exploring how protein aggregation interacts with other pathways, including mitochondrial dysfunction, ROS, and inflammation. These interactions are critical in determining disease progression.
Finally, advances in imaging and biomarker development are improving the ability to detect protein aggregation earlier in patients, which may enable diseases to be diagnosed sooner and more targeted treatment to be utilized.
Disease Connections
Explore the clinical manifestations where this specific biological mechanism plays a primary role in neurodegeneration and disease progression.
Sources
Dobson, C. M. (2003). Protein folding and misfolding.
Knowles, T. P. J., Vendruscolo, M., & Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases.
Taylor, J. P., Brown, R. H. Jr., & Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism.
Soto, C., & Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases.
Ross, C. A., & Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease.
Ciechanover, A., & Kwon, Y. T. (2015). Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies.